Introduction
On 31 December 2019, China reported a cluster of cases of pneumonia in people from Wuhan, Hubei Province. The responsible pathogen was a novel coronavirus, named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It is the cause of COVID-19, a disease which can result in death associated with acute respiratory distress syndrome (ARDS), usually secondary to pneumonia.
By 16 May 2020, there were 4,555,803 people infected worldwide, with 304,201 deaths. The estimated damage to the world economy for 2020 is $1 trillion. At least $5 trillion has been allocated by first world countries as part of an economic rescue package for their businesses and workers.
There are intensive international efforts to discover a vaccine to SARS-CoV-19. A commercially available vaccine may not be through phase 3 trials until mid-2021.
Against the background of this viral pandemic, currently available medications are under investigation as treatment options for COVID-19. One of these is tocilizumab (TCZ).
Tocilizumab
Tocilizumab (Actemra®) is indicated for rheumatoid arthritis (with methotrexate, if possible), juvenile idiopathic arthritis (systemic or polyarticular – with methotrexate, if possible), giant cell arteritis (GCA), with adjunct corticosteroid treatment, and in cytokine release syndrome induced by chimeric antigen receptor T cell (CAR-T) therapy. It is available as 80 mg in 4 mL, 200 mg in 10 mL and 400 mg in 20 mL.
Tocilizumab is a humanised monoclonal antibody (IgG). It binds to the interleukin-6 (IL-6) receptors throughout the body leading to rapid reductions in erythrocyte sedimentation rate (ESR) and concentrations of C-reactive protein (CRP). Tocilizumab may increase the activity of hepatic enzyme 1A2, 2C9, 2C19 and 3A4, thereby potentially affecting the metabolism of other drugs.
In the AMBITION study, most adverse reactions were mild or moderate, with less than 3.8% of patients experiencing severe adverse reactions. Skin infections were the most common, but less than the rate seen in patients taking methotrexate. Liver function tests were elevated, but without signs or symptoms of hepatitis or hepatic dysfunction. Mouth ulceration and gastritis have also been reported, and some patients have suffered perforation of the gut, mainly secondary to diverticulitis.
Lung histology
A brief review of lung histology is beneficial to the understanding of the link between IL-6 and SARS- CoV-2’s lethality.
The alveolar epithelium is composed of three types of cells:
- Type I cells which comprise 90% of the alveolar surface area,
- Type II cells which are approximately 10% of the area, and contain the ACE2 receptor
- Macrophages
Type 1 cells are responsible for gas exchange, whilst type II cells produce a surfactant which lowers the cell surface tension at the air-liquid (inhaled water) interface, thereby preventing the alveoli from collapsing together. Type II cells also remove excess alveolar fluid through intracellular transport and contain the angiotensin-converting enzyme 2 (ACE2) receptor.
Macrophages are the most numerous immune cells present in the lung environment under homeostatic conditions. They are integral to the innate defence of the airways. They exhibit a malleability of function which covers the maintenance of pulmonary homeostasis, microbial clearance, removal of cellular debris, immune monitoring, responses to infection and the resolution of inflammation.
The role of interleukin 6 (IL-6)
Inflammation is a core initiator of the innate immune system and is the body’s first line of defence against infection or injury. Inflammation also activates the adaptive (or acquired) immune system.
Interleukin-6 is classified as a pro-inflammatory cytokine and is produced predominantly by activated macrophages, but also T cells, endothelial cells, fibroblasts and hepatocytes. IL-6 exhibits a duality of functionality – it also responds to neutrophils and monocytes/macrophages (part of the innate immune system). This duality can cause amplification of inflammation and a switch from an acute to a chronic inflammatory state.
IL-6 is considered one of the most important cytokines released during a viral infection and aids host defence by the up-regulation of acute phase responses such as C-reactive protein (CRP), serum amyloid A (SAA), fibrinogen, and haptoglobin, and decreases the levels of fibronectin, albumin and transferrin. IL-6 has a stimulatory effect on T- and B-cells (part of the adaptive/acquired immune system). In particular, “IL-6 promotes specific differentiation of naïve CD4+ T cells, thus performing an important function in linking the innate and acquired immune response.”
The production of IL-6 is tightly controlled, and cytokine down-regulation is vital because dysregulated production of IL-6 has a deleterious effect on both host autoimmunity and inflammation. For example, elevated IL-6 levels reduce zinc levels, and can induce autoantibody production, thrombocytosis and hypergammaglobulinaemia. Also, excessive levels of SAA cause amyloid fibril deposits, resulting in gradual multi-organ deterioration, whilst increased hepcidin levels reduce serum iron levels, causing hypoferremia and anaemia.
The down-regulation of the IL-6 mediated inflammatory response is effected by IL-10, an anti-inflammatory cytokine, as well as transforming growth factor β (TGF-β). The reduction in IL-6 activity is necessary so as to prevent the “cytokine storm” – a lethal state of systemic inflammation.
SARS-CoV-2, immune response and the cytokine storm
When SARS CoV-2, an enveloped, single-stranded, positive-sense RNA virus is inhaled, it gains entry into the host cell in a two-stage process.
First, the viral spike (S) protein binds to the ACE-2 receptor, located on type-II cells. This facilitates viral attachment. The S protein is then further primed for fusion and entry into the host cell by the protease, TMPRSS2.
As a result of this binding and host invasion by SARS-CoV-2, both the innate and the adaptive immune systems are activated, with the consequent release of numerous types of cytokines including IL-6. This constitutes a functional immune response.
However, in severe COVID-19 patients, the immune response becomes dysregulated, with a pathological effect on both inflammation and autoimmunity. In particular, there are elevated levels of IL-6 which contributes to the cytokine storm, an immune system over-reaction.
IL-6 levels in COVID-19 patients have been reported to exceed 627.1pg/mL before TCZ therapy. The normal level is 5pg/mL or less.
Aside from elevated IL-6 levels, severe COVID-19 patients also have lower levels of beneficial CD4+ and CD8+ T cells, both of which are part of the adaptive (or acquired) immune system.
This effect of IL-6 confirms that the cytokine storm diminishes adaptive immunity against SARS-CoV-19 infection, thus making the patient less likely to recover. As Liu, et al. (2020) has noted, “The more serious the disease and the worse the prognosis, the lower were the T cell, CD4+ T cell, and CD8+ T cell counts on admission. Based on these findings, we believe that the CD4+ and CD8+ T cell counts in patients with COVID-19 could reflect disease severity and predict disease prognosis and are therefore good biomarkers of COVID-19 activity.”
In short, IL-6 shifts from acting to protect the host due via a regulated immune system, to becoming a threat to host survival due to a dysregulated immune system.
Mechanism of the cytokine storm
The replication and release of the virus cause inflammatory host cell death (pyroptosis). Damage-associated molecular patterns (DAMP) are recognised by alveolar macrophages which start the production of pro-inflammatory cytokines and chemokines including IL-6. The initial action of IL-6 is beneficial to the host as it contributes to the immune response. However, as further monocytes, macrophages and T cells are attracted to the infection site, due to the increasing number of viral particles, there is an augmentation of the inflammation process. A pro-inflammatory cytokine “feedback loop” is created.
The dysregulated consequences associated with the increase in IL-6 include enhanced vascular endothelial permeability, resulting in increased levels of neutrophils in the lungs. Neutrophil entry into the lungs is a defining feature of ARDS. Neutrophils cause an increase in capillary permeability, resulting in alveolar oedema and arterial hypoxaemia, further compromising ongoing gas exchange and adding to the onset of ARDS. ARDS mortality corresponds to the degree of neutrophilia in the lung. There is also a diffuse thickening of the alveolar wall, further decreasing gas exchange.
Respiratory failure caused 69.5% of deaths in one Wuhan hospital cohort of 82 patients.
Another consequence of neutrophil-induced capillary permeability is that pro-inflammatory IL-6 can escape into the systemic circulation. The systemic ‘escape’ of supraphysiological levels of IL-6 is implicated in multi-organ damage, including kidney disease, cardiovascular disease including fulminant myocarditis, and hepatic failure. The systemic viral damage occurs because IL-6 receptors are ubiquitous. Foetal tissue studies show IL-6 receptors not only in the lungs but also the eye, heart, liver, spleen, and adrenal, renal and gastrointestinal tissue.
Adding to the consequences of the cytokine storm is clinical evidence from human and animal studies indicating that an “overexpression of IL-6 might be a possible mechanism favouring persistence of some viruses”, perhaps secondary to the reduced levels of CD4+ and CD8+ T cells noted earlier.
The multi-factorial interplay of immune components, cytokines and SARS-CoV-2 is well represented in Figure 1.
Figure 1: Cytokine storm and T cell lymphopenia is associated with COVID-19 severity (reproduced with permission from Pedersen et al.)
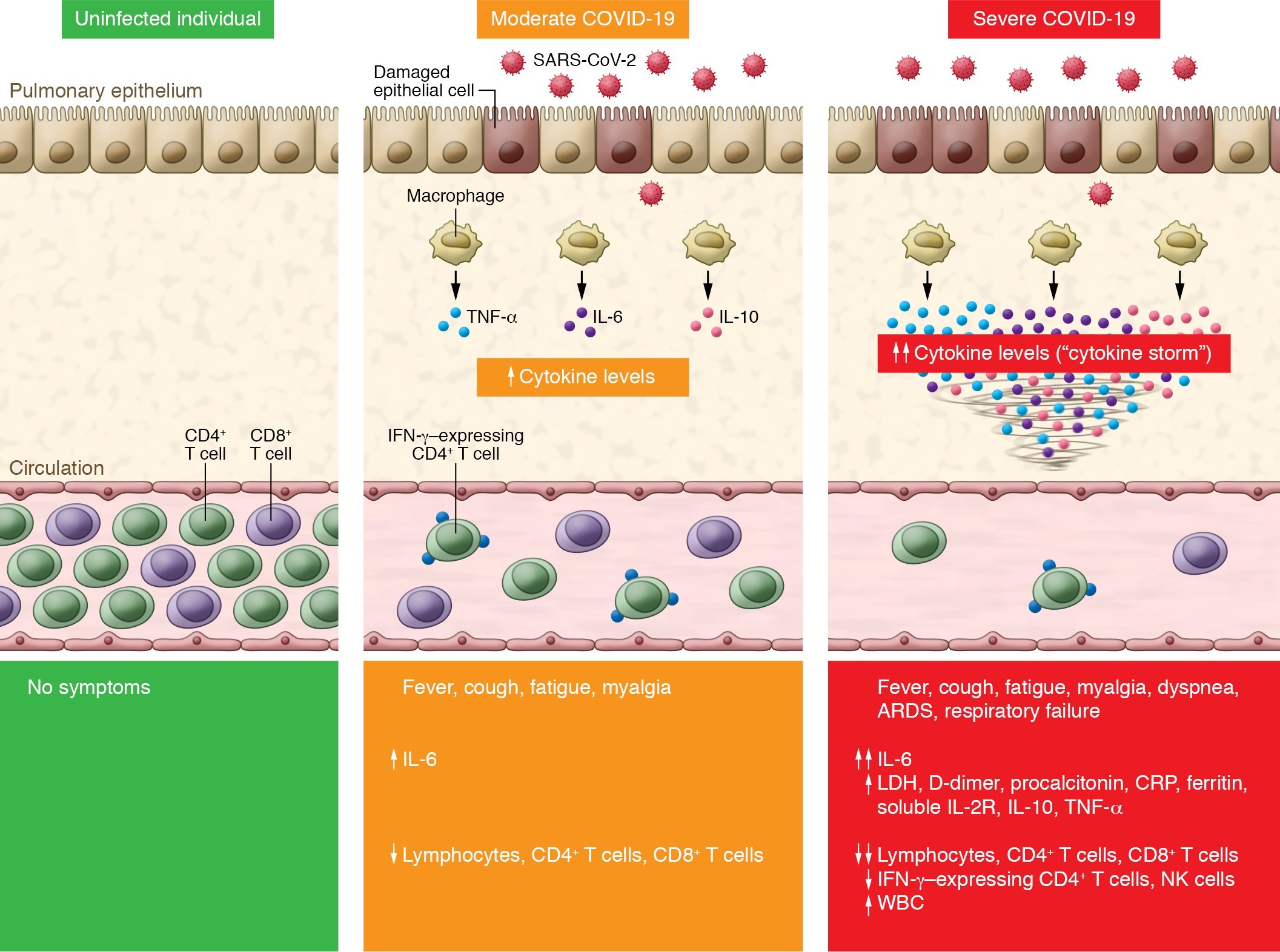
SARS-CoV-2 infection causes COVID-19. Compared with uninfected individuals (left panel), moderate COVID-19 cases exhibit an increase in IL-6 and a decrease in total T lymphocyte counts, particularly CD4+ T cells and CD8+ T cells (middle panel). Severe COVID-19 cases have further increased production of IL-6, IL-2R, IL-10, and TNF-α, while total T lymphocytes, particularly CD4+ T cells and CD8+ T cells, and IFN-γ–expressing CD4+ T cells markedly decrease (right panel). The level of cytokine storm and T cell lymphopenia is associated with pulmonary damage, respiratory distress, and unfavourable outcome. ARDS, acute respiratory distress syndrome; CRP, C-reactive protein; LDH, lactate dehydrogenase.
SARS-CoV2, cardiovascular complications and ACE2 receptors.
Research indicates that there are several prognostic indicators for severe disease and ICU admission in COVID-19 patients. These include being elderly and having underlying comorbidities such as chronic obstructive pulmonary disease (COPD), cardiovascular disease and hypertension. One study has reported that 21% of COVID-19 patients had hypertension. Hypertension is associated with a nearly 2.5-fold significantly increased risk of severe COVID-19 disease and a similar significantly higher risk of mortality. Cardiovascular morbidity – characterised by tachyarrhythmias, elevated troponin, and thromboembolic events – is reported in 20% of hospitalised patients and is strongly allied with mortality risk.
The elucidated explanation for the higher mortality rate in hypertensive patients is due to the destruction of the ACE2 receptors after SARS-Cov2 binding to the type II cells and subsequent cell lysis.
Importantly, ACE2 receptors are also located on the heart, gastrointestinal tract and kidneys at higher levels than are found in the lungs.
The physiological function of ACE2 is to convert angiotensin II to angiotensin 1-7. The ACE2-angiotensin 1-7 axis is a counterbalancing arm to the ACE1-angiotensin II arm of the renin-angiotensin system (RAS).
Angiotensin II can induce vasoconstriction and possesses pro-inflammatory and profibrotic capacities. In contrast, angiotensin 1-7 is antiproliferative, antiapoptotic and has vasodilating actions. It is also cardioprotective in its capacity to be anti-heart failure, antithrombotic, anti-myocardial hypertrophy, antifibrosis, antiarrhythmic and anti-atherogenic.
As can be appreciated, with the destruction of ACE2 receptors on type II cells, the delicate balance between ACE1 and ACE2 can lead to dysregulation of blood pressure and other deleterious cardiovascular sequelae.
The role and interplay between SARS-CoV2, the RAS and higher mortality rates are elegantly portrayed in Figure 2.
Figure 2. The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2)/severe acute respiratory syndrome coronavirus (SARS‐CoV) infection could possibly influence the balance between angiotensin II (Ang II) and angiotensin 1‐7 (Ang‐[1‐7]) (from Guo et al.)
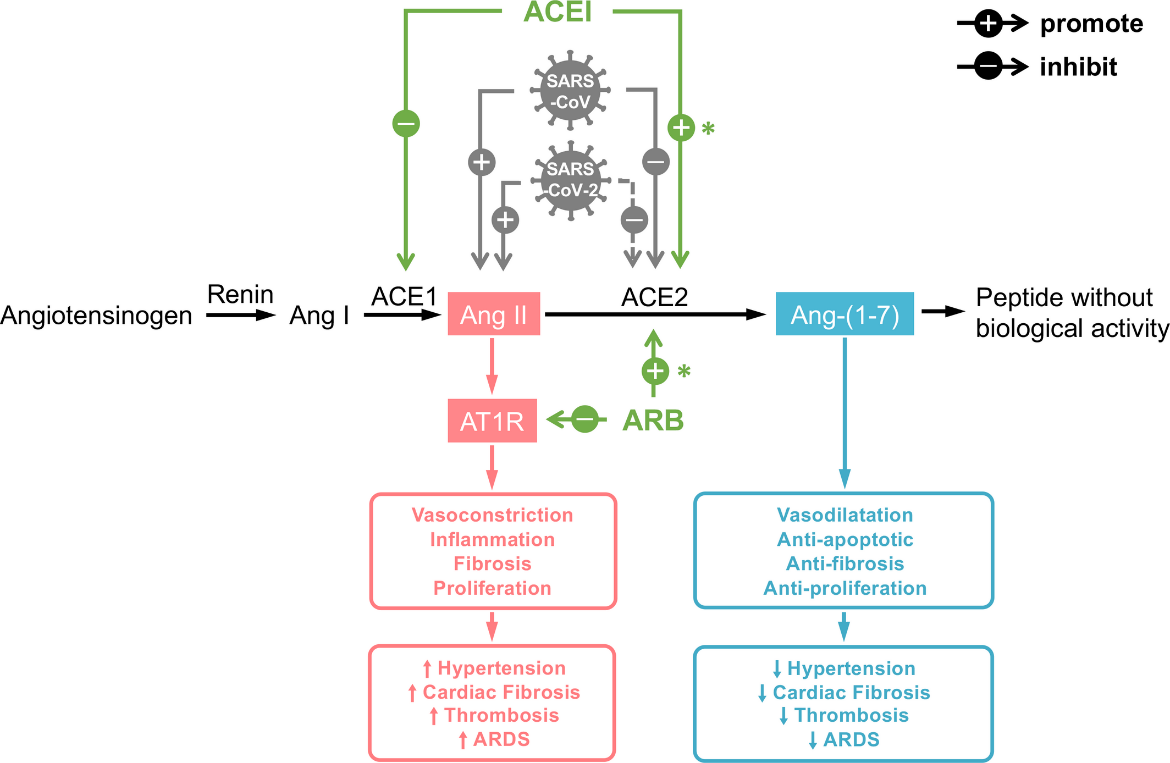
*indicates finding in hearts; ACE1, angiotensin‐converting enzyme 1; ACE2, angiotensin‐converting enzyme 2; ACEI, angiotensin‐converting enzyme inhibitor; Ang I, angiotensin I; ARB, angiotensin receptor blocker; ARDS, acute respiratory distress syndrome; AT1R, angiotensin II type 1 receptor; dotted line, speculation based on the current evidence; solid line, findings from current evidence; up arrow, promote; down arrow, inhibit.
SARS-CoV-2 and stroke in COVID-19 patients
On 28 April 2020, the New England Journal of Medicine reported on five COVID-19 patients who presented with large-vessel stroke. A possible explanation may involve virus-induced destruction of the ACE-2 receptor and reduced AT1-7 generation, the production of superoxides causing resultant endothelial damage, and the putative impact of this cascade on the release of von Willebrand factor (vWF) and the link to clot formation and stroke. The release of vWF is also triggered by IL-6.
Kerr (2001) reported that IL-6 promotes coagulation via an increased transcription of Factor VIII as well as causing a 4.5-fold increase in tissue factor mRNA. IL-6 also increases both platelet production and activation, and vWF. Similarly, Chin (2003) has reported that “elevated IL-6 may contribute to the thrombotic and thromboembolic complications in acute heart failure, in a process mediated via increased TF [tissue factor] and vWF.”
Peyvandhi (2011) and Escher (2020) have concluded that vWF plays a pivotal role in primary haemostasis by facilitating platelet adhesion to damaged vascular endothelium and subsequent platelet aggregation. High levels of vWF, in part caused by elevated IL-6 levels, are predictive of endothelial activation, disease outcome and future prothrombotic events.
Trials supporting TCZ in COVID patients
Fu et al. (2020) reported on a trial of TCZ in 21 severe or critical COVID-19 patients with elevated IL-6 levels of greater than 20pg/mL. Patients were receiving standard treatments including lopinavir, methylprednisolone, other supportive treatments and oxygen. To recall, the normal level of IL-6 is 5pg/mL or less.
Patients were given a first dose of TCZ at 4-8mg/kg (recommended 400mg), diluted to 100mL with 0.9% normal saline and infused over one1 hour. For poor responders, the same dose as the first was given 12 hours later. No single dose over 800mg was given.
Twenty of twenty-one patients recovered and were discharged within two weeks, whilst one patient was slower to be discharged.
The researchers concluded that “Tocilizumab treatment is recommended to reduce the mortality of severe COVID-19.”
It is important to note that there may be a transient increase in IL-6 levels lasting a few days after administration of TCZ because the IL-6 receptor is now blocked.
Michot and colleagues (2020), in a journal pre-proof case publication have reported on an immunocompromised patient with renal cell carcinoma, who tested positive for SARS-CoV-2. A chest computerised tomography (CT) scan revealed bilateral patchy ground glass opacities. Lopinavir-ritonavir (400mg-100mg orally) was commenced on day seven for five days. On day eight, sudden dyspnoea occurred with a saturation drop requiring oxygen at 6L/min, but without artificial ventilation.
The patient was given two doses of TCZ, 8mg/kg IV, eight hours apart. He became rapidly afebrile, with good clinical improvement and was slowly weaning off oxygen. A day 12 chest CT showed partial regression of the pulmonary infiltrates and ground glass appearance. His C-reactive protein (CRP), a surrogate marker for cytokine storm has reduced from 225mg/L to 33mg/L in four days.
The reference range normal for CRP is less than 5mg/L, and a CRP greater than 10mg/L is indicative of an acute infection or inflammation.
Luo et al. (2020) conducted a retrospective study of 15 COVID-19 patients. Two patients were moderately ill, six were seriously ill, and seven were critically ill. Eight patients received TCZ with methylprednisolone (MPS). Five patients received two or more dose of TCZ. Four of the critical patients only received one dose of TCZ 320-600mg plus MPS 40mg -160mg across 3-5 days.
Before TCZ therapy, the CRP level in deceased patients 1,2 and 3 ranged from 175.8mg/L to 257.9mg/L and reduced to 12.8-51mg/L prior to death.
Interestingly patient 6, who survived, had a pre-TCZ CRP of 253.1 mg/L which reduced to 5.0mg/L on day 7.
The pre- and post-TCZ IL-6 levels followed the predicted pattern of an increased level post-TCZ in 14 of the 15 patients. Some post-TCZ levels were dramatic. For example, patient 4’s level increased from 392 pg/mL pre-TCZ to 935.5 pg/mL one day after TCZ but reduced to 396.8 pg/mL on day 4.
Also, the pre-TCZ IL-6 level was not always predictive of outcome. Patients 1, 2 and 3 had pre-TCZ IL-6 levels of 16.4, 32.7 and 73.6pg/mL respectively. At the time of death, the IL6 levels ranged between 2230.0 – 5000.0pg/mL. In comparison, patient 14 had a pre-TCZ IL-6 level of 627.1pg/mL which reduced to 249.0 pg/mL on day 7.
Whilst this study has many weaknesses, including variable doses of variable drugs and low study numbers, the overall conclusion by the authors was that in critically ill patients, or those with a high IL-6 titre, two or three doses would be clinically indicated.
The 29 April 2020 pre-publication information from the CORIMUNO-TOCI French study of 129 COVID-19 patients with medium to severe pneumonia suggest that there was a significant reduction in the proportion of patients who had to be transferred to resuscitation units or who died. Half of the patients received one or two injections of TCZ plus standard care (oxygen, antibiotics and anticoagulants) whilst the other half received only standard care. Follow-up was 14 days. Importantly TCZ was effective in preventing the “inflammatory storm.”
Conclusion
Because there is little expectation of a vaccine to SARS-CoV-2 before 2021, there is a pressing, indeed urgent need for the safe and prudent use of currently available medications. The mechanism of action of tocilizumab theoretically suggests, and early research confirms, that it is a suitable candidate in moderate-severe COVID-19 patients.