

The Therapeutic Goods Administration (TGA) has issued an alert advising of new prescribing restrictions for hydroxychloroquine. These restrictions have been introduced following reports of increased off-label prescribing for the management of coronavirus disease (COVID-19), which have led to concerns of supply interruptions.
Several small studies suggest that hydroxychloroquine and the similar agent chloroquine (not marketed in Australia) could be effective against COVID-19. Interest in chloroquine for COVID-19 may stem from a study that found the agent to be effective in preventing the replication of coronavirus (SARS-CoV) in vitro. However, the evidence for effectiveness in COVID-19 is considered limited and clinical trials are continuing around the world. Clinical trials are also set to begin in Australia following announcements by the University of Queensland and the Walter and Eliza Hall Institute in Melbourne.
To prevent stock shortages related to a sudden increase in demand, the TGA has amended the Poisons Standard. As of 24 March 2020, hydroxychloroquine can only be initiated by medical practitioners specialising in the following fields:
- Dermatology;
- Intensive care medicine;
- Paediatrics and child health;
- Physician; or
- Emergency medicine.
Other medical practitioners can continue to prescribe repeats for hydroxychloroquine for patients initiated on therapy prior to 24 March 2020, in line with the registered indications. Hydroxychloroquine is currently registered in Australia for the treatment of rheumatoid arthritis, mild systemic and discoid lupus erythematosus, and the suppression and treatment of malaria.
The Australian Technical Advisory Group on Immunisation (ATAGI) has released its advice for the administration of seasonal influenza vaccines in 2020. Influenza vaccines used in Australia this year will be quadrivalent and comprise two A strains (H1N1 and H3N2) and two B strains. The influenza vaccines registered for use in 2020 are shown in Table 1.
Table 1. Influenza vaccines registered for use in Australia in 2020
| Product |
Type |
Registered age group |
| FluQuadri® |
Split virion |
Adults and children ≥ 6 months |
| Vaxigrip® Tetra |
Split virion |
Adults and children ≥ 6 months |
| Fluarix® Tetra |
Split virion |
Adults and children ≥ 6 months |
| Influvac® Tetra |
Surface antigen |
Adults and children ≥ 3 years |
| Afluria® Quad |
Split virion |
Adults and children ≥ 5 years |
| Fluad® Quad |
Surface antigen, adjuvanted |
Adults ≥ 65 years |
Significant changes for the 2020 season include:
- All children aged six months to less than five years are eligible to receive an influenza vaccine funded through the National Immunisation Program (NIP);
- The registered age range for FluQuadri® has been extended to include children from six months of age. FluQuadri® Junior is no longer available;
- The registered age range for Influvac® Tetra has been extended to include children from three years of age; and
- The dose for all influenza vaccines is now 0.5mL regardless of patient age.
With the novel threat of coronavirus disease (COVID-19), the influenza vaccination program may be particularly important this year in reducing the risk of severe illness and hospitalisation. Information on patient eligibility under the NIP can be obtained from the Department of Health. Individual product information documents should be consulted for further details.


A generic tiotropium capsule has recently been added to the Pharmaceutical Benefits Scheme (PBS). Braltus® capsules are considered equivalent to Spiriva® capsules for the purposes of brand substitution on the PBS. However, some important differences should be noted to avoid confusion.
Braltus® capsules for inhalation contain 13 micrograms of tiotropium, while Spiriva® contains 18 micrograms per capsule. However, the dose delivered from the mouthpiece of the inhaler device is 10 micrograms per capsule for each brand. Braltus® is able to provide the same delivered dose from a smaller formulated dose due to the enhanced aerosolisation properties of the formulation.
Braltus® capsules should only be administered using the Zonda® inhaler device included in each pack. While the Zonda® device is operationally similar to the Spiriva® HandiHaler® device, the devices are not interchangeable. Patients should be educated on proper inhaler technique for each device they use.
A comparison of the two brands of tiotropium capsules is shown below.
| |
Braltus® |
Spiriva® |
| Active ingredient |
Tiotropium (as bromide) |
Tiotropium (as bromide monohydrate) |
| Tiotropium per capsule |
13mcg |
18mcg |
| Tiotropium delivered dose per capsule |
10mcg |
| Recommended dose |
1 capsule inhaled daily |
| Indications |
Long-term maintenance treatment of bronchospasm and dyspnoea associated with COPD; prevention of COPD exacerbations |
| Device |
Zonda® |
HandiHaler® |
| Device maintenance |
Discard device after 30 uses. New device supplied with each pack. |
Device can be washed and re-used |
| Presentation |
Bottle, clear capsules |
Blister pack, green capsules |

Topical corticosteroids have long been the mainstay of treatment for dermatitis that does not respond to moisturisers and emollients. Crisaborole is a new non-steroidal option approved for use in mild to moderate atopic dermatitis in patients two years of age and older. Although its precise mechanism of action is not well defined, crisaborole inhibits phosphodiesterase-4 (PDE-4) which leads to reduced secretion of pro-inflammatory cytokines such as tumour necrosis factor-alpha (TNFα).
Crisaborole ointment is applied as a thin layer to the affected areas twice daily. The ointment can be used on all skin areas, including the face and intertriginous areas. Two large randomised, vehicle-controlled clinical trials demonstrate the efficacy of crisaborole. These studies show a significant improvement in disease severity from day eight of treatment and an early and sustained improvement in pruritus in patients treated with crisaborole compared to the vehicle-treated group. The efficacy of crisaborole has not been studied beyond a 28-day treatment course in a controlled trial. However, an open-label extension trial suggests that multiple treatment courses over an extended period may be an effective treatment option that avoids the potential concerns of long-term corticosteroid therapy.
Approximately 25% of the crisaborole dose is absorbed following topical administration. However, the potential for systemic effects is limited as crisaborole is rapidly and substantially metabolised into inactive metabolites. Application site pain was the only treatment-related adverse event associated with crisaborole therapy in two pivotal trials.


Pembrolizumab is now available on the Pharmaceutical Benefits Scheme (PBS) for the first-line treatment of metastatic non-small cell lung cancer (NSCLC) irrespective of programmed death ligand-1 (PD-L1) status.
In the KEYNOTE-189 trial, pembrolizumab was used in conjunction with pemetrexed and platinum chemotherapy in metastatic NSCLC irrespective of PD-L1 status, opening up a new standard of care for these patients. The median duration follow-up was 18.7 months, and pembrolizumab was associated with a significantly longer overall survival (22.0 versus 10.7 months) compared to pemetrexed and platinum-based chemotherapy alone.
The incidence of treatment-related adverse effects was similar between the treatment arms. However, immune-mediated adverse effects including hypothyroidism and hyperthyroidism, pneumonitis, skin toxicity, colitis and infusion reactions, were markedly higher with the pembrolizumab combination at 22.7% and chemotherapy at 11.9%.
An updated edition of the Therapeutic Guidelines: Oral and Dental has recently been released. This resource covers a broad range of conditions that are relevant to both dentists and medical practitioners.
Some of the significant changes include:
- An expanded guide to the common causes and appropriate management of acute dental pain;
- Clarification of the indications for antibiotic therapy;
- Addition of photos to assist recognition of common disease of the oral mucosa;
- Addition of information relating to appropriate treatment modification for patients with complex medical conditions;
- Expansion of information on medication-related osteonecrosis of the jaw to assist risk assessment and the consequent management of at-risk patients;
- Inclusion of advice for the management of patients at risk of adrenocortical insufficiency;
- Addition of a table summarising the properties of different local anaesthetics along with example calculations of the maximum dose;
- Expanded information on the safe use of anxiolysis in the perioperative period. A printable patient information sheet has also been included;
- Addition of a new section involving the management of peri-implant disease; and
- Expansion of information to assist medical practitioners in the management of common oral and dental conditions.
If you are currently using an offline form of the Therapeutic Guidelines, it is recommended to download the updated version at your earliest convenience.
New contraindications have been added to the product information of Ganfort® (bimatoprost + timolol). It is now advised that these eye drops not be used in patients with sino-atrial nodal block or second or third-degree atrioventricular block not controlled with a pacemaker.
Timolol is a non-selective beta-blocker. Systemic effects of timolol may include bradycardia, orthostatic hypotension, syncope, and bronchoconstriction. While it may sometimes be assumed that eye drops do not produce significant systemic effects, studies suggest that at least 80% of an ophthalmic product drains through the nasolacrimal canal. From there, it may enter the systemic circulation while also avoiding first-pass metabolism.
The current contraindications for Ganfort® eye drops include:
- Bronchospasm;
- Bronchial asthma;
- Severe chronic obstructive pulmonary disease;
- Sinus bradycardia;
- Sick sinus syndrome;
- Sino-atrial nodal block;
- Second or third-degree atrioventricular block not controlled with a pacemaker;
- Overt cardiac failure; and
- Cardiogenic shock.
To avoid systemic absorption, it is recommended to immediately apply pressure to the tear duct for at least two minutes following administration of the eye drop. This is good practice for all eye drops to minimise systemic effects, avoid an unpleasant taste in the mouth, and improve retention at the site of action.
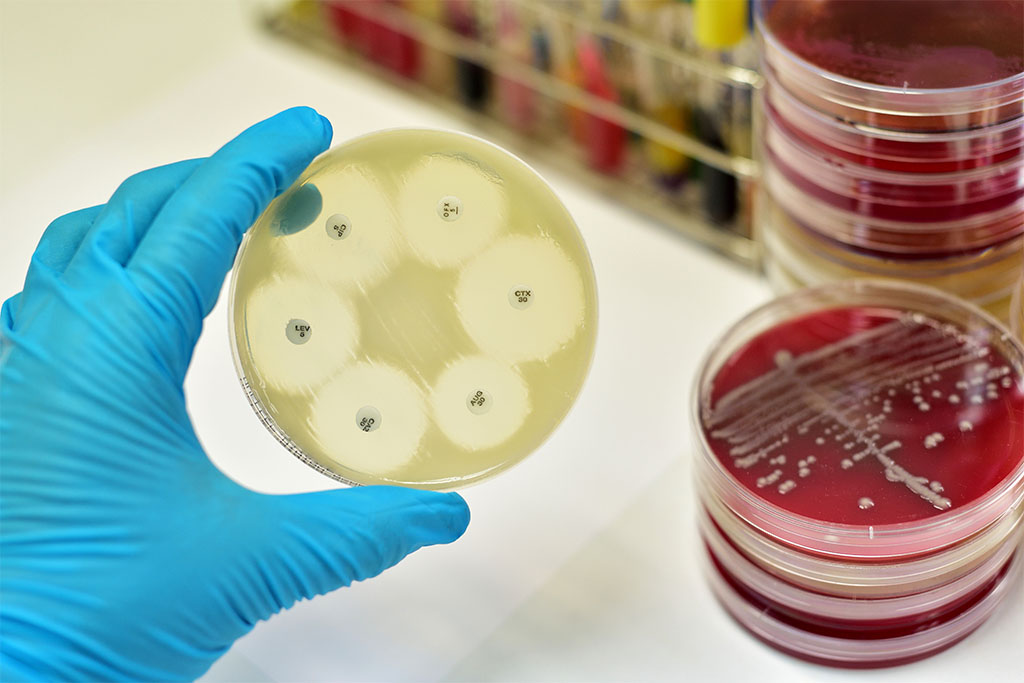
Zavicefta® is a new injectable cephalosporin formulation available in Australia. This product combines the broad-spectrum antibiotic, ceftazidime with the β-lactamase inhibitor, avibactam.
Ceftazidime is one of a limited range of antibiotics with antipseudomonal activity. However, the 2019 AURA Report (Antimicrobial Use and Resistance in Australia) demonstrates that resistance amongst Pseudomonas aeruginosa to ceftazidime has increased from 4.4% of isolates in 2014 to 5.1% in 2017. Avibactam inhibits many of the bacterial enzymes known to inactivate ceftazidime. This extends its spectrum of activity to cover a variety of multi-drug resistant Gram-negative bacteria. However, the formulation has little or no activity against most Gram-positive organisms and anaerobes. Additional agents should be used when such pathogens are known or suspected to be contributing to the infectious process.
Zavicefta® is indicated for the treatment of complicated intra-abdominal infection (in combination with metronidazole), complicated urinary tract infection, and hospital-acquired pneumonia. Ceftazidime and avibactam are both excreted into the urine unchanged; dose adjustment is recommended in moderate and severe renal impairment. The most commonly reported adverse effects are nausea and diarrhoea, which is usually mild to moderate in severity. Clostridium difficile-associated diarrhoea has been reported and should be considered if diarrhoea occurs during, or several weeks after finishing, therapy.
Broad-spectrum antibiotics such as ceftazidime increase the risk of colonisation with antibiotic-resistant microorganisms. They should, therefore, be used judiciously.


Teduglutide is a glucagon-like peptide (GLP) analogue recently registered in Australia for the treatment of short bowel syndrome. There are a number of other GLP analogues available including dulaglutide, exenatide, and liraglutide. While these agents are GLP-1 analogues, teduglutide is an analogue of GLP-2.
The glucagon-like peptides, GLP-1 and GLP-2, are naturally secreted in equivalent amounts but can have very different effects in the body. For example, GLP-1 significantly reduces the levels of the lipoprotein chylomicron, while GLP-2 increases its levels. Other effects of GLP-1 include the promotion of glucose-dependent insulin secretion, preservation of pancreatic β-cell function, slowing of gastric emptying, and reduced appetite. These agents have, therefore, been used in the management of obesity and type 2 diabetes. On the other hand, GLP-2 increases intestinal and portal blood flow, inhibits gastric acid secretion, and slows intestinal motility. These effects have led to the use of teduglutide in the treatment of short bowel syndrome in patients who are dependent on parenteral support.
Clinical studies demonstrate that teduglutide promotes normal growth and repair throughout the intestines. Citrulline, an amino acid produced predominantly by enterocytes, can be considered a marker of enterocyte mass. Over a 24-week study, teduglutide increased plasma citrulline by 20.6µmol/L compared to a 0.7µmol/L increase in the placebo group. Patients in the teduglutide group enjoyed a mean reduction in weekly parenteral support of 4.4L compared to 2.3L in the placebo group.
Adverse effects most commonly reported in clinical trials include abdominal pain and distension, nausea, and injection site reactions. Owing to its pharmacological activity, it is thought that teduglutide may be able to produce hyperplastic changes in the small bowel and hepatobiliary tract. Although this has not been demonstrated in clinical studies, patients should be monitored and therapy discontinued in cases of active gastrointestinal malignancy.
Neratinib is a new tyrosine kinase inhibitor for the treatment of breast cancer. It is indicated as extended adjuvant treatment of early-stage human epidermal growth factor receptor (HER) 2 -positive disease. Initiation is recommended to occur within 12 months of completing trastuzumab therapy.
Tyrosine kinases are important proteins involved in cellular signalling and have a range of biological actions. Members of the HER family of tyrosine kinases promote tumour cell proliferation and survival. Neratinib is known as a pan-HER inhibitor as it has activity at HER1, HER2, and HER4 receptors. Inhibition of these receptors prevents activation of the signal transduction pathway, leading to apoptosis and reduced cellular proliferation. Unlike other tyrosine kinase inhibitors, such as lapatinib, neratinib binds to its target irreversibly.
Diarrhoea is a very common adverse effect, reported in 93.6% of patients. It is recommended that anti-diarrhoeal prophylaxis be initiated with the first dose and continue during the first two months of treatment. Neratinib dose adjustment may be required with permanent discontinuation recommended in severe cases.
Neratinib has a number of clinically important drug interactions. As it is primarily metabolised via cytochrome P450 3A4, exposure can be significantly affected by inducers or inhibitors of this hepatic enzyme. It is contraindicated with strong inducers (e.g. carbamazepine, phenytoin, St John’s wort, and rifampicin) and moderate inhibitors (e.g. fluconazole, diltiazem, verapamil, and erythromycin). Drugs that alter the pH of the upper gastrointestinal tract may also affect therapy as neratinib requires an acidic environment for absorption.