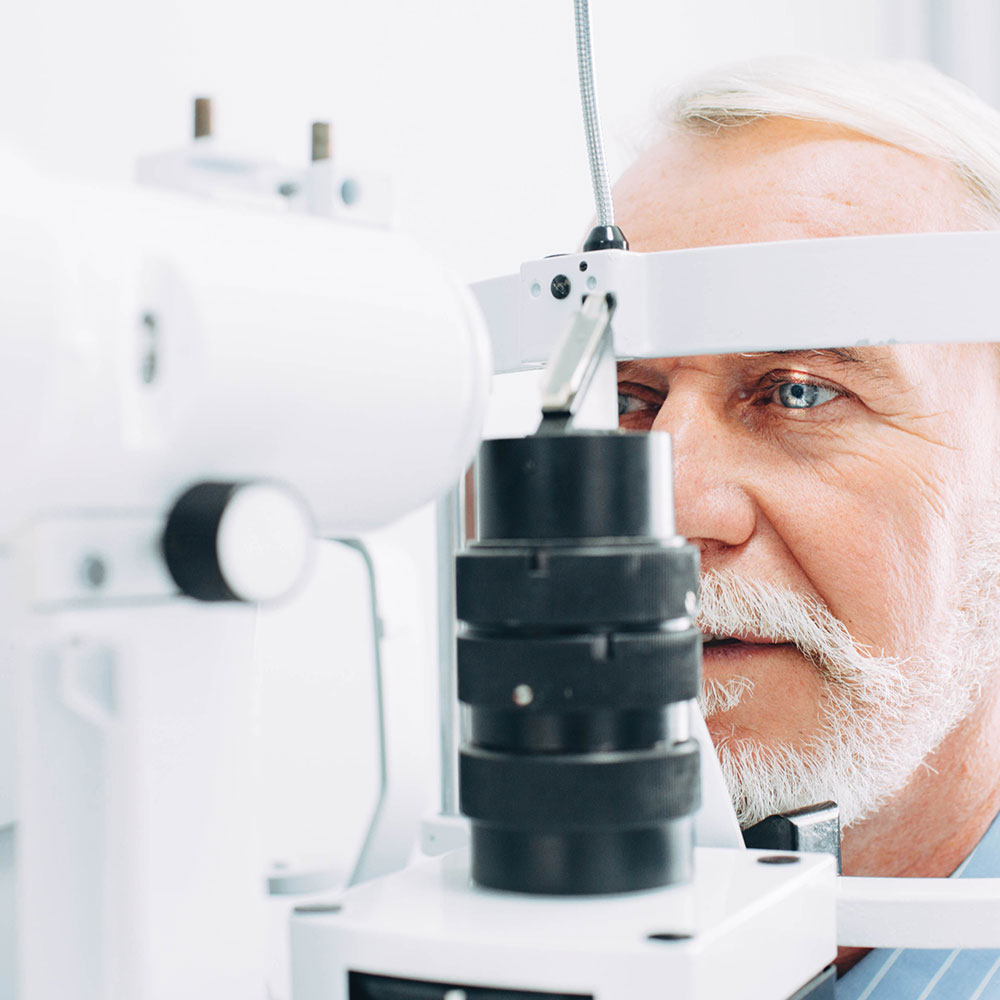
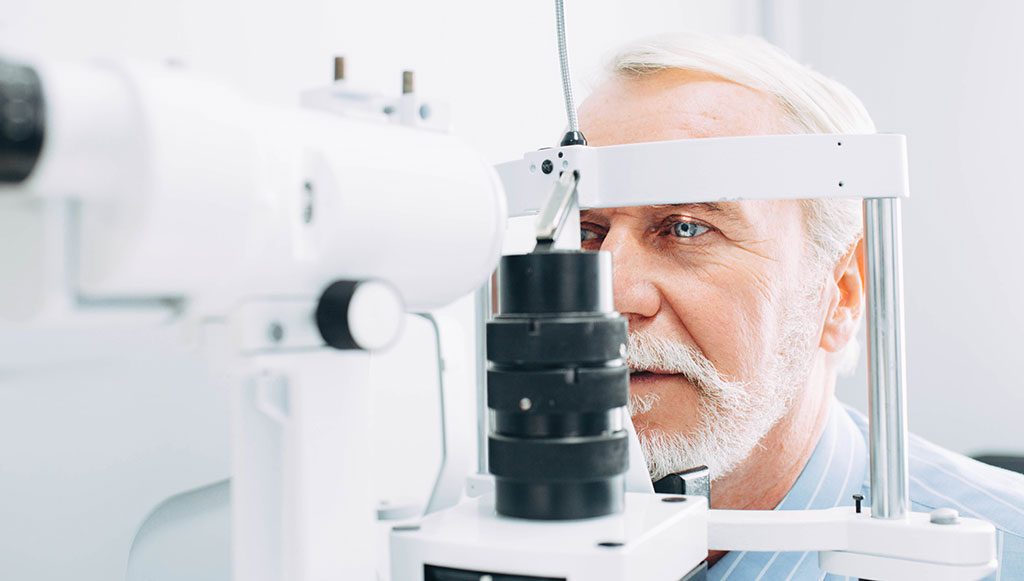
The Pharmaceutical Benefits Scheme (PBS) listing for aflibercept was expanded on 1 October 2020. Aflibercept is now subsidised for the treatment of subfoveal choroidal neovascularisation, in addition to macular oedema secondary to diabetes or retinal vein occlusion.
Aflibercept binds vascular endothelial growth factor-A (VEGF-A) and placental growth factor (PlGF) to prevent their interaction with VEGF receptors. Excessive activation of VEGF receptors is associated with pathologic angiogenesis and increased vascular permeability. Interruption of these pathways by aflibercept can slow vision loss by reducing vascular leakage and neovascularisation.
Aflibercept is administered by ophthalmologists as an intravitreal injection. Following administration, patients must be monitored for increased intraocular pressure. Patients should also be advised to seek immediate medical advice if they experience any symptoms indicative of endophthalmitis such as eye pain or redness, photophobia, or blurred vision.


Lorlatinib was recently added to the Pharmaceutical Benefits Scheme (PBS) for the treatment of metastatic non-small cell lung cancer (NSCLC). To be eligible for subsidised therapy, patients must have evidence of an anaplastic lymphoma kinase (ALK) gene rearrangement and have experienced disease progression following treatment with an ALK inhibitor other than crizotinib.
Lorlatinib is a tyrosine kinase inhibitor with activity against ALK and the structurally similar protein, c-ros oncogene 1 (ROS1) kinase. Inhibition of these kinases impairs signalling pathways involved in cell proliferation and survival in lung and other cancers. ALK inhibitors, such as crizotinib, alectinib, and ceritinib, have improved the prognosis for ALK-positive tumours. However, relapse and disease progression are common due to acquired resistance.
Lorlatinib is a third-generation ALK inhibitor that overcomes some of the resistance mechanisms of other agents in this class. It also demonstrates improved penetration of the blood-brain barrier compared to crizotinib, which provides better intracranial disease control.
In a Phase I/II study, the most commonly reported adverse events included hypercholesterolaemia, hypertriglyceridaemia, oedema, peripheral neuropathy, cognitive effects, and diarrhoea. Serious adverse events were reported in just over a third of the cohort, and 2.7% of patients permanently discontinued lorlatinib due to treatment-related adverse events.


From 18th September 2020, all products containing bufexamac will be removed from the Australian Register of Therapeutic Goods (ARTG). This action follows an investigation by the Therapeutic Goods Administration (TGA), which concluded that the risk-benefit ratio of bufexamac is not favourable.
Bufexamac is a non-steroidal anti-inflammatory drug (NSAID) and is a well-documented cause of allergic contact dermatitis. Reactions range from localised irritation to widespread polymorphic eruptions, with some patients requiring hospitalisation. The risk of developing a reaction to bufexamac is high, particularly in atopic individuals. Bufexamac has, therefore, been withdrawn in many countries where it was once approved for the treatment of dermatitis. In Australia, bufexamac was only found in combination with a topical anaesthetic and antiseptic for the first aid treatment of minor skin complaints such as abrasions and stings.
In addition to the risk of allergic reactions, the TGA declares that there is insufficient evidence to support the efficacy of bufexamac. While bufexamac will no longer be sold in Australia, a range of alternative first aid creams remain available.
On September 1, acalabrutinib was listed on the Pharmaceutical Benefits Scheme (PBS) for the treatment of relapsed or refractory chronic lymphocytic leukaemia (CLL) and small lymphocytic lymphoma (SLL). Acalabrutinib is an inhibitor of Bruton’s tyrosine kinase (BTK), an important enzyme in oncogenic signalling. Inhibition of BTK ultimately leads to reduced proliferation and survival of malignant B-cells.
The efficacy of acalabrutinib in the treatment of CLL was assessed in two randomised Phase III clinical trials. In the ASCEND trial, patients received acalabrutinib monotherapy or physician’s choice (rituximab plus idelalisib or bendamustine) with a median follow-up of 16.1 months. The median progression-free survival (PFS) was not reached in the acalabrutinib group, compared to 16.5 months for the control group. In the ELEVATE-TN trial, patients received acalabrutinib monotherapy, acalabrutinib with obinutuzumab, or obinutuzumab with chlorambucil. After a median follow-up of 28.3 months, the median PFS was not reached for patients taking acalabrutinib, compared to 22.6 months for the control group.
The most common adverse events reported during these clinical trials include anaemia, neutropenia, thrombocytopenia, upper respiratory tract infection, and diarrhoea. Bleeding events, such as bruising and petechiae, are very common; major haemorrhage was reported in 3.6% of patients. To minimise the risk of bleeding, physicians may consider withholding therapy in the three to seven days before and after surgery.
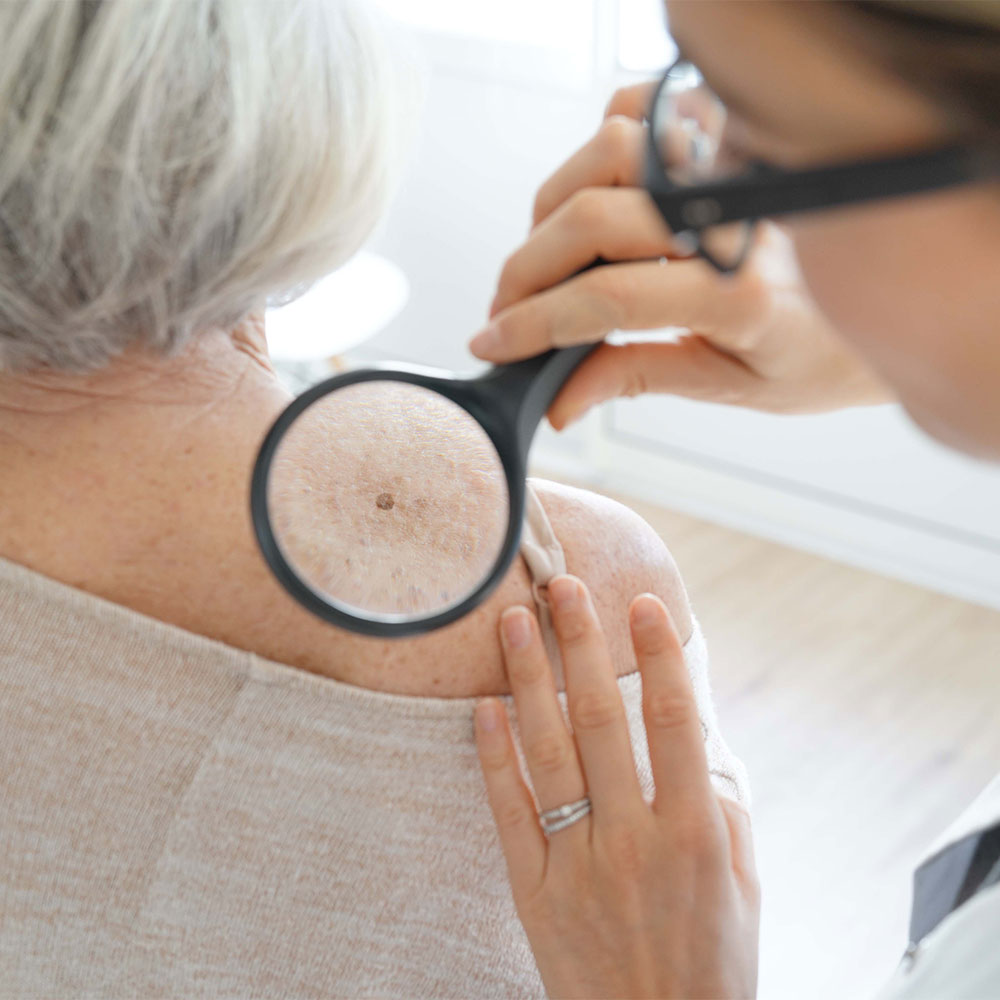
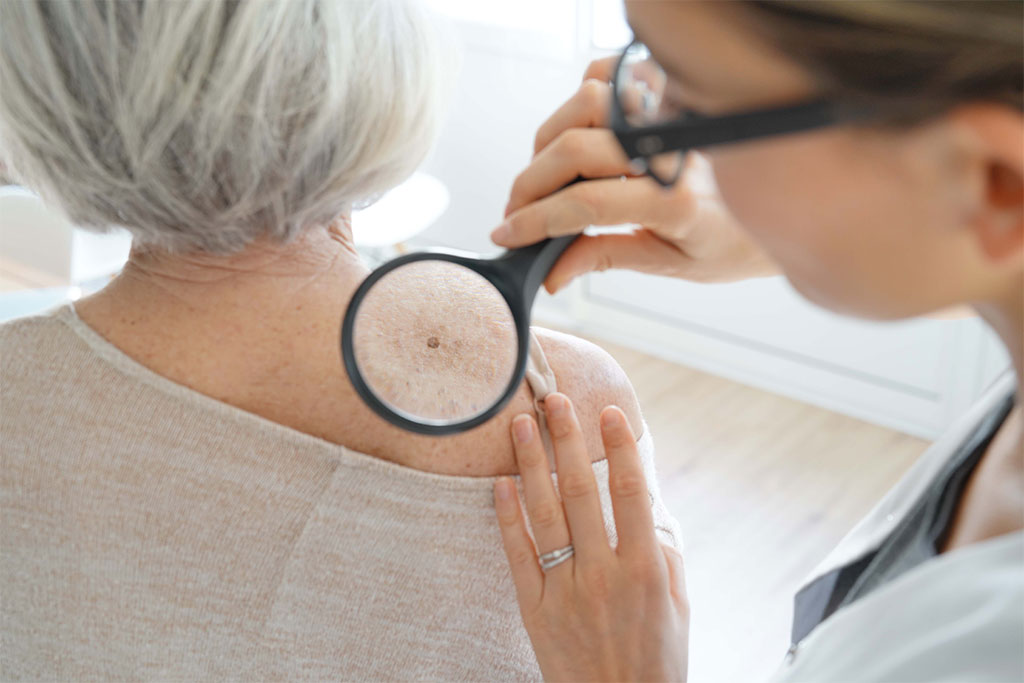
Evidence suggests that hydrochlorothiazide is associated with an increased risk of carcinoma in sun-exposed tissues. It is theorised that this may be due to the photosensitising properties of hydrochlorothiazide.
A recent Australian study demonstrated that hydrochlorothiazide use was associated with a 2.6-fold greater risk of lip cancer and a 20% increased risk of cutaneous melanoma. The risk also appeared to be dose-related, as patients with high hydrochlorothiazide use (defined as a cumulative dose of ≥ 25,000mg) demonstrated a 4.7-fold higher odds of lip cancer. This echoes the findings of two Danish case-control studies that showed an increased risk of squamous cell carcinoma and basal cell carcinoma with hydrochlorothiazide use. This increased risk was not observed for furosemide, angiotensin-converting enzyme (ACE) inhibitors, or angiotensin II antagonists.
Hydrochlorothiazide is a thiazide diuretic indicated for the treatment of hypertension and oedema. It is available as a single ingredient tablet and is also formulated with other antihypertensive agents in a range of fixed-dose combination products. Patients taking hydrochlorothiazide should be advised to regularly check their skin and lips and promptly report any new or changed skin lesions or moles to their doctor. Patients should also limit their sun exposure and practice sun safety.
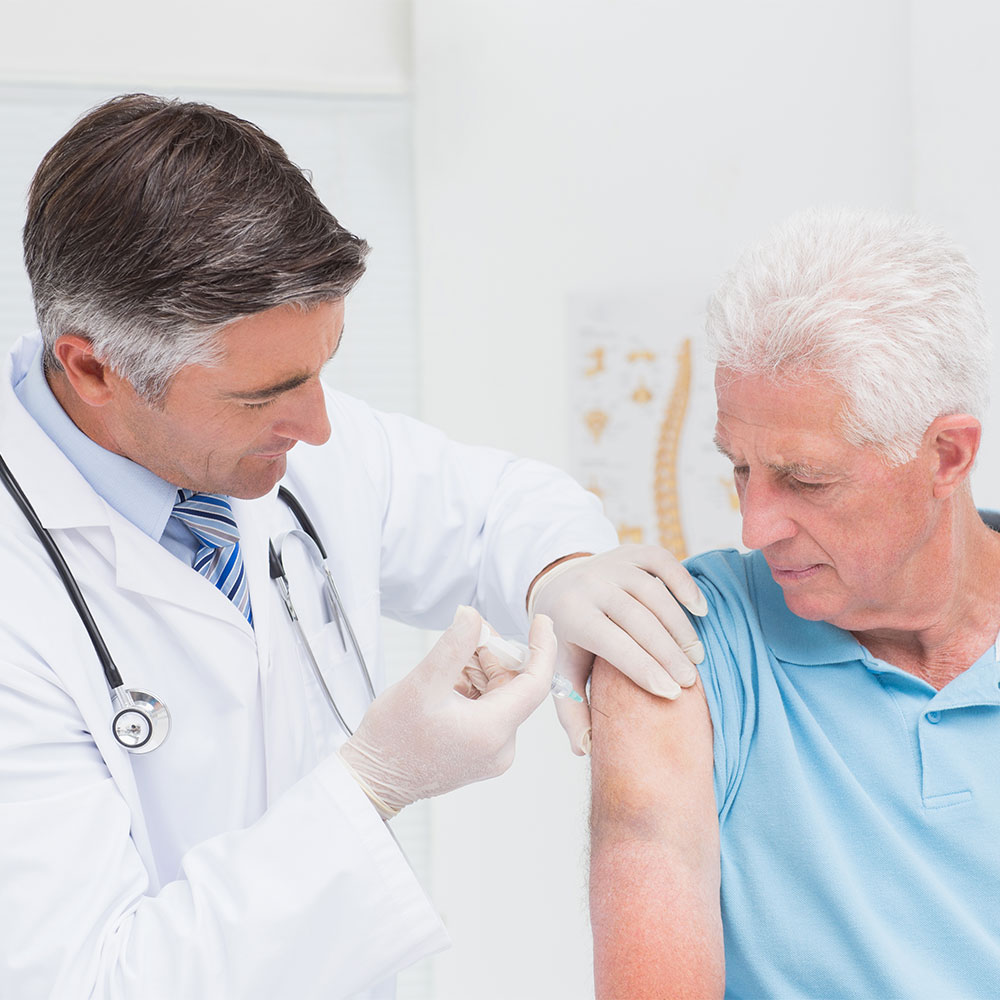
The Therapeutic Goods Administration (TGA) has issued a safety advisory regarding the use of Zostavax® in people with compromised immune function. Zostavax® is a live, attenuated varicella-zoster vaccine indicated for the prevention of herpes zoster in people aged 50 years and older. This vaccine is contraindicated in people with primary and acquired immunodeficiency states as well as those receiving immunosuppressive therapy. However, the product information emphasises that it is not contraindicated in people receiving topical or inhaled corticosteroids, low-dose systemic corticosteroids, or corticosteroid replacement therapy.
The TGA warning follows a new report of death due to disseminated varicella-zoster virus (Oka vaccine strain) infection. This patient was taking hydroxychloroquine and low-dose prednisolone when they received the zoster vaccine, in line with clinical recommendations. The TGA reminds healthcare professionals that this potentially life-threatening condition can occur in patients on low-dose immunosuppressive therapy and highlights the importance of a risk-based assessment prior to Zostavax® administration.
The Australian Immunisation Handbook can be referred to for further information regarding zoster vaccine use in people taking immunosuppressive therapy and the value of serological testing.


Semaglutide is the latest glucagon-like peptide-1 (GLP-1) analogue to be added to the Pharmaceutical Benefits Scheme (PBS). GLP-1 is an incretin hormone that stimulates the release of insulin and inhibits the release of glucagon in a glucose-dependent manner. Semaglutide is indicated for the treatment of type 2 diabetes mellitus.
In a 30-week double-blind clinical trial, the mean baseline glycated haemoglobin (HbA1c) level fell from 68mmol/mol (8.4%) to 48mmol/mol (6.5%) in the group receiving semaglutide 1mg weekly, compared to 67mmol/mol (8.3%) in the placebo group. A target HbA1c of <53mmol/mol (7.0%) was achieved in 79% of the semaglutide 1mg group compared to 11% of the placebo group.
Semaglutide is administered weekly as a subcutaneous injection. Gastrointestinal adverse effects such as nausea, vomiting, and diarrhoea are commonly experienced. These effects are usually mild to moderate in severity and reduce as therapy continues. Weight loss is commonly observed due to reduced appetite. Body weight reductions of ≥5% may be achieved in around two-thirds of patients, while losses of ≥10% occur in a quarter of patients. A reduction in the dose of concomitant insulin or sulfonylurea therapy may be required when starting semaglutide to reduce the risk of hypoglycaemia. Pancreatitis has been associated with the use of GLP-1 analogues. While the risk is thought to be low, patients should be counselled to seek immediate medical attention if they experience unexplained severe abdominal pain.
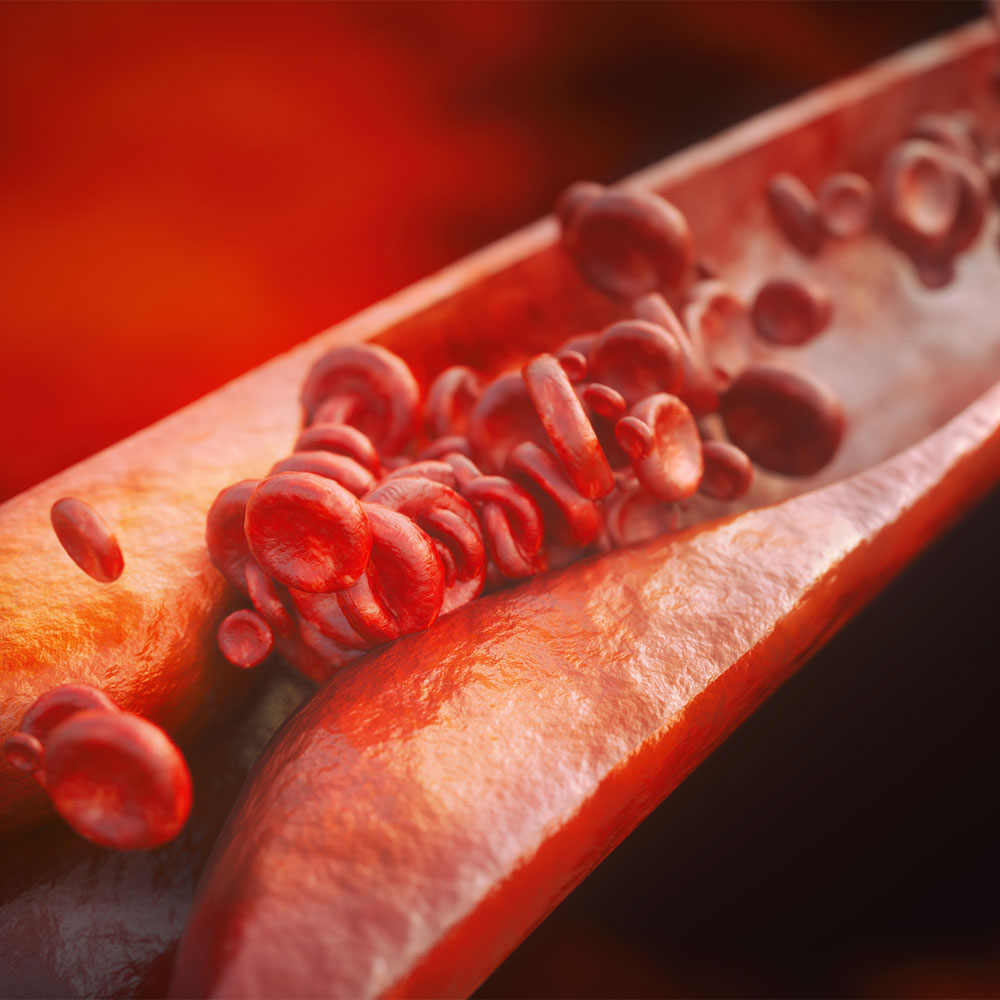
Evolocumab is now available on the Pharmaceutical Benefits Scheme (PBS) for high-risk atherosclerotic cardiovascular disease (ASCVD) in non-familial hypercholesterolaemia.
The PBS criteria must include low-density lipoprotein (LDL) levels >2.6mmol/L, symptomatic ASCVD with additional risk factors and the patient must have had ≥ 12 weeks of optimised therapy.
In the FOURIER trial, evolocumab was demonstrated to significantly reduce LDL cholesterol levels by 59% compared to placebo.
Evolocumab significantly reduced the risk of the primary composite endpoint (hazard ratio 0.85) of cardiovascular death, myocardial infarction, hospitalisation for unstable angina or coronary revascularisation, as well as key secondary composite endpoint (hazard 0.80) of cardiovascular death, myocardial infarction or stroke.
Adverse effects include injection side reactions, nausea, back or joint pain, nasopharyngitis, the common cold, flu-like symptoms, and in rarer situations, hypersensitivity reactions including angioedema.


Trifluridine/tipiracil is now available on the Pharmaceutical Benefits Scheme (PBS) for pre-treated cancer of the stomach or gastro-oesophageal junction.
In the TAGS trial, trifluridine/tipiracil was demonstrated to significantly prolong overall survival in pre-treated gastric cancer (5.7 months vs 3.6 months) compared to placebo, with 21% of treated patients alive at 1 year.
The disease control rate was 44% for patients who had a complete or partial response to trifluridine/tipiracil, where time of deterioration of ECOG (Eastern Cooperative Oncology Group) performance status almost doubled (4.3 months vs 2.3 months) compared to placebo.
The incidence of treatment-related adverse effects was consistent with that seen for metastatic colorectal cancer, with haematological adverse effects markedly higher, including neutropenia, anaemia, leucopenia and thrombocytopenia.


The Pharmaceutical Benefits Scheme (PBS) has recently expanded the listing criteria for Symbicort®. Symbicort® contains the corticosteroid, budesonide plus the long-acting β2-agonist (LABA), formoterol.
The new PBS listing allows Symbicort® to be used as reliever therapy in the management of mild asthma in patients 12 years of age and older who are not currently using a single agent LABA. Previously, Symbicort® was only PBS listed for the treatment of moderate to severe forms of asthma, as well as chronic obstructive pulmonary disease (COPD).
This new PBS listing aligns with the recommendations of the Global Initiative for Asthma which no longer recommends the use of a short-acting β2-agonist alone for adults and adolescents with asthma. Instead, an inhaled corticosteroid plus LABA is their preferred option for symptom relief in these patients. In patients with mild asthma, randomised controlled trials demonstrate that this therapy is associated with a 64% lower rate of severe exacerbations compared to treatment with a short-acting β2-agonist alone.
The following table shows the PBS streamlined authority codes for the different presentations of Symbicort®. Please refer to the PBS website for full clinical criteria.
| Product |
PBS Authority Streamlined Codes |
| Presentation |
Strength |
Mild asthma |
Asthma |
COPD |
| Symbicort® Rapihaler® |
50mcg/3mcg |
|
4397 |
|
| 100mcg/3mcg |
10482 |
4397 |
|
| 200mcg/6mcg |
|
4404 |
10121 |
| Symbicort® Turbuhaler® |
100mcg/6mcg |
|
4380 |
|
| 200mcg/6mcg |
10464 |
7970 |
|
| 400mcg/12mcg |
|
7979 |
10121 |