

Palbociclib has recently listed on the Pharmaceutical Benefits Scheme (PBS) for the treatment of locally advanced or metastatic breast cancer. Palbociclib is a selective inhibitor of cyclin-dependent kinases (CDKs) 4 and 6. Cyclins and their complex interactions with associated kinases are important drivers of the mammalian cell cycle. Dysregulation of these processes frequently occurs in breast cancer, leading to uncontrolled cellular proliferation.
The double-blind, phase 3 PALOMA-2 study investigated the efficacy of palbociclib with letrozole compared to placebo plus letrozole. The progression-free survival (PFS) was 24.8 months in the palbociclib-letrozole group compared to 14.5 months in the placebo-letrozole group. Follow-up is ongoing to allow assessment of overall survival. The rate of Grade 3-4 adverse events was higher in the palbociclib-letrozole group, with 9.7% of patients permanently discontinuing treatment (compared to 5.9% in the placebo-letrozole group). The most common Grade 3 and 4 events reported in the palbociclib-letrozole group were neutropenia and leukopenia at 66.4% and 24.7% respectively, compared to 1.4% and 0% for the placebo-letrozole group.
Palbociclib is the second cyclin-dependent kinase (CDK) inhibitor to be listed for breast cancer following the approval of ribociclib in July 2018. The PBS conditions for palbociclib are similar to those of ribociclib in that the condition must be hormone receptor (HR)-positive and human epidermal growth factor receptor 2 (HER2)-negative and treatment must be in combination with anastrozole or letrozole. Studies suggest that palbociclib has similar efficacy to ribociclib in terms of PFS delay, overall response rate, and time to deterioration in quality of life. Their spectrum of adverse effects is also similar, although palbociclib does not appear to prolong the QTc interval.


Fiasp® is now available on the Pharmaceutical Benefits Scheme (PBS) for the treatment of diabetes mellitus. Fiasp® is a novel formulation of insulin aspart containing the excipients nicotinamide (vitamin B3) and arginine hydrochloride.
Human insulin molecules naturally self-associate to form hexamers that are too large to readily cross capillary membranes. The rate-limiting step for absorption and onset of action following subcutaneous injection is, therefore, the conversion of these hexamers into smaller units. Nicotinamide increases the rate of dissociation of insulin hexamers into dimers and monomers, resulting in faster absorption. Arginine is an amino acid that has been added to stabilise the formulation.
As nicotinamide and arginine hydrochloride are considered excipients, the generic name for Fiasp® is simply insulin aspart. It is important to note that this formulation is not interchangeable with NovoRapid®. In comparison to NovoRapid®, Fiasp® has an onset of action that is around five minutes faster and a glucose-lowering effect that is 74% greater during the first 30 minutes. Clinical trials demonstrate that this results in improved postprandial blood glucose control without increasing the overall risk of hypoglycaemia. The total and maximum glucose-lowering effect of Fiasp® is similar to that of NovoRapid®.
To avoid confusion, the Australian Commission on Safety and Quality in Healthcare recommend all insulin orders specify the full brand name of the product to be administered.


The European Medicines Agency (EMA) has announced prescribing restrictions of alemtuzumab for multiple sclerosis (MS) while a safety review is undertaken. This action is in response to new reports of serious cardiovascular and immune-mediated adverse events including heart attack, stroke, cervicocephalic arterial dissection, neutropenia, and autoimmune hepatitis.
Alemtuzumab is a monoclonal antibody active against the CD52 cell surface antigen. This protein is present on T and B lymphocytes at high levels and to a lesser extent on natural killer cells, monocytes, and macrophages. Alemtuzumab is thought to improve relapsing forms of MS through the depletion and repopulation of lymphocytes. A higher strength of alemtuzumab (MabCampath®) is also registered in Australia for the treatment of B-cell chronic lymphocytic leukaemia.
The Therapeutic Goods Administration (TGA) has not announced any action at this stage. Actions recommended by international governing bodies such as the EMA and the US Food and Drug Administration (FDA) for patients with MS include:
- Consider ceasing therapy in patients who develop signs of the abovementioned adverse events;
- Advise patients to seek immediate medical attention if they experience any symptoms of these adverse events;
- Monitor vital signs before and during alemtuzumab infusions;
- Perform liver function tests before and during therapy;
- Promptly evaluate patients who develop signs of pathological immune activation; and
- Consider other treatment options for new patients.
These serious adverse events are thought to be rare. A total of four strokes and no heart attacks have been reported to the TGA in association with both alemtuzumab formulations since the drug was first approved in Australia in 2006.


Safinamide has recently been added to the Pharmaceutical Benefits Scheme (PBS). It is indicated for the treatment of fluctuating idiopathic Parkinson’s disease as an adjunct to a levodopa-decarboxylase inhibitor combination.
Safinamide is a highly selective and reversible inhibitor of monoamine oxidase B (MAO-B). It works to increase extracellular levels of dopamine in the striatum; a region of the brain responsible for voluntary movement. Safinamide also inhibits the release of glutamate which may have additive benefits for the management of motor complications. Clinical trials demonstrate that safinamide increases daily on time without troublesome dyskinesia by up to 0.55 hours compared to placebo.
The selectivity of safinamide for MAO-B is more than 1,000 times greater than its selectivity for MAO-A. Therefore, dietary tyramine restrictions are not required. However, other medications that inhibit MAO activity must not be used with safinamide. Caution should be exercised when serotonergic agents are used concomitantly; pethidine use is contraindicated. Safinamide may also transiently inhibit the multi-drug transporter known as breast cancer resistance protein (BCRP). At least five hours should be allowed between safinamide doses and BCRP substrates such as pravastatin, ciprofloxacin, methotrexate, topotecan, and diclofenac.
The most common treatment-emergent adverse events reported in clinical trials are dyskinesia, falls, nausea, and insomnia. Retinal degeneration was observed in some animal studies. Although this effect was not reported in human trials, safinamide should be avoided in patients at higher risk of potential retinal effects. Contraindications include albino patients, retinal degeneration, uveitis, inherited retinopathy, and severe progressive diabetic retinopathy.
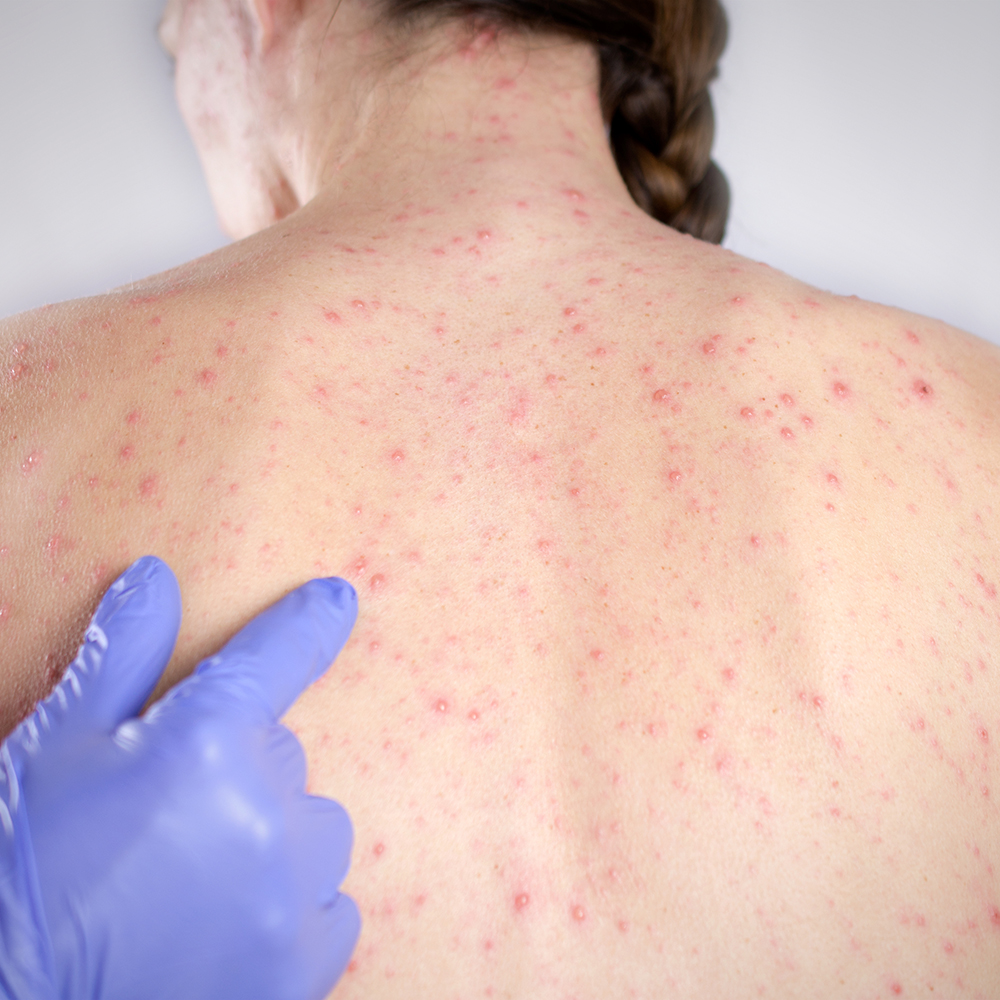
In 2014, the World Health Organization (WHO) declared measles to be eliminated in Australia. While no longer endemic, imported cases of measles still occur which can lead to localised outbreaks. A total of 108 measles cases have been reported to the National Notifiable Diseases Surveillance System in Australia so far this year compared to 103 for the whole of 2018.
Measles has an estimated basic reproduction number (R0) in the range of 12-18. This means that an average of 12 to 18 secondary infections are expected following one typical case in a totally susceptible population. Due to the highly contagious nature of this virus, Australia has an immunisation coverage target of 95%.
It is recommended that individuals receive two doses of measles-containing vaccine, with the first dose administered at 12 months of age. However, a recent update to the Australian Immunisation Handbook advises that the first dose may be given to infants as young as six months of age in special circumstances. Special circumstances include travel to highly endemic areas, during outbreaks, and as post-exposure prophylaxis.
A recent study suggests that infants vaccinated against measles between six and eight months of age have lower long-term immunity to measles compared to infants who receive their first dose between nine and 12 months of age. At 14 months of age, 20% of children in the early vaccination group had antibody concentrations below the cut-off for clinical protection compared to 0% in the group vaccinated later. This effect persisted for the duration of the study with 11.1% of the early vaccinated group vaccinated early not achieving clinically protective antibody levels three years after their booster compared to 0% of the group vaccinated later. The Australian Immunisation Handbook advises that infants who receive an early dose of measles-containing vaccine should receive two additional doses on the recommended schedule.

Humalog® U200, a double strength formulation of insulin lispro, has recently been added to the Pharmaceutical Benefits Scheme (PBS). This ultra-short-acting insulin is indicated for the management of type 1 and type 2 diabetes mellitus.
The range of products containing insulin lispro is shown in Table 1.
Table 1. Insulin lispro products available
| Product |
Insulin |
Strength |
Presentation |
| Humalog® |
Insulin lispro |
100units/mL |
Vial, cartridge, KwikPen® device |
| Humalog® U200 |
Insulin lispro |
200units/mL |
KwikPen® device |
| Humalog Mix25® |
Insulin lispro + lispro protamine |
100units/mL |
Cartridge, KwikPen® device |
| Humalog Mix50® |
Insulin lispro + lispro protamine |
100units/mL |
Cartridge, KwikPen® device |
This new high-strength formulation is only available in the disposable KwikPen® device. The prescribed dose is dialled up in units for both the 200 units/mL and the 100 units/mL KwikPen® device. Therefore, no dose conversion is required when a patient changes to the new strength. To avoid dosing errors, the solution from the pre-filled pen must not be mixed with any other insulin or transferred to any other insulin delivery device. Drawing insulin out of the Humalog U200 KwikPen® and measuring the dose using an insulin needle with unit markings would result in a two-fold dosing error.
Insulin is considered high-strength when the concentration exceeds 100 units/mL. High-strength insulins such as Humalog® U200 and Toujeo® (insulin glargine 300 units/mL) may be beneficial for patients needing high daily doses as lower injection volumes are required. This can reduce the pain of injection and may also limit variability in absorption.
The Therapeutic Goods Administration (TGA) has issued a safety alert for tofacitinib following identification of an increased risk of pulmonary embolism and death in a post-marketing study. This safety signal was detected in a cohort of rheumatoid arthritis patients taking 10mg twice daily. These results have not been observed in other clinical trials or post-marketing data.
Tofacitinib is a Janus kinase inhibitor indicated for the treatment of rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis. Tofacitinib is only approved at a dose of 5mg twice daily for the treatment of rheumatoid arthritis. However, higher doses are approved for treatment induction of ulcerative colitis.
Patients who were taking tofacitinib 10mg twice daily have transitioned to lower doses for the remainder of the study. The sponsor is continuing to work with the TGA to investigate this issue and will provide further details as they come to light. In the interim, healthcare professionals are reminded to follow the recommendations in the product information in relation to the specific condition for which tofacitinib is prescribed. Patients should be advised to seek urgent medical attention if they experience symptoms such as:
- Sudden shortness of breath or difficulty breathing;
- Pain in the chest or back;
- Coughing up blood;
- Excessive sweating; or
- Clammy/bluish coloured
Healthcare professionals and patients are encouraged to report any adverse events or side effects associated with the use of tofacitinib to the TGA.


Around 63% of Australian adults are overweight or obese, significantly increasing their risk of disease such as cardiovascular disease, type 2 diabetes, and some cancers. Contrave® is a new medication for weight management as an adjunct to diet and exercise. It is indicated for adults who are obese and adults who are overweight and have at least one weight-related comorbidity. Contrave® contains the opioid antagonist, naltrexone, in combination with the dopamine and noradrenaline reuptake inhibitor, bupropion.
A randomised, double-blind, placebo-controlled trial demonstrates the efficacy of this combination therapy. Weight loss of at least 5% was achieved in 31% of participants taking bupropion with naltrexone compared to 12% in the placebo group. Weight loss of at least 10% and 15% was observed in 17% and 9% of the treatment group respectively (5% and 2% for placebo). Significant improvements were also observed in waist circumference, insulin resistance, and lipid levels.
In clinical trials, twice as many people discontinued Contrave® therapy due to adverse events compared to placebo. The most commonly reported reactions leading to discontinuation were nausea, headache, dizziness, and vomiting. As bupropion is associated with a dose-related risk of seizures, Contrave® is contraindicated in patients with a history of seizures. Caution should also be exercised in patients with risk factors for seizures such as a history of head trauma, excessive alcohol use, use of medications that may induce hypoglycaemia, and concomitant use of drugs that may lower the seizure threshold.
Carbimazole is an anti-thyroid medication indicated for the treatment of hyperthyroidism. Two safety issues have recently been highlighted following a review by the European Medicines Agency (EMA):
- Risk of pancreatitis; and
- The importance of contraceptive measures.
Post-marketing surveillance reveals an association between acute pancreatitis and carbimazole therapy. Although the mechanism for this effect is poorly understood, it is thought to have an immunological basis. If acute pancreatitis occurs, carbimazole should be discontinued immediately and not restarted. Re-exposure may result in life-threatening acute pancreatitis with a reduced time to onset.
Pancreatitis may present with fever, nausea, vomiting, and abdominal pain that is usually central and epigastric; plasma lipase levels are often at least three times greater than the reference range. While a number of case reports have been published on this issue, only one report of pancreatitis with carbimazole can be found in the almost 50 years of data available on the Australian Database of Adverse Event Notifications. It is also worth noting that carbimazole was not the only medication suspected of causing the adverse event in this case.
Available data now also strengthens the evidence that carbimazole can cause congenital malformations when administered during pregnancy. Reported malformations include aplasia cutis congenita, craniofacial malformations, exomphalos, oesophageal atresia, omphalo-mesenteric duct anomaly, and ventricular septal defect. The risk appears to be greatest during the first trimester and when high doses are used. It is, therefore, recommended that women of childbearing potential use effective contraceptive measures while undertaking therapy with carbimazole.
Studies also demonstrate that the risk of congenital malformations is greater when maternal hyperthyroidism remains untreated. Carbimazole should only be used during pregnancy after a careful individual risk/benefit assessment. If carbimazole is deemed appropriate, the lowest effective dose should be prescribed without additional administration of thyroid hormones. Close maternal, foetal, and neonatal monitoring is recommended.
The manufacturer has advised that the product information for carbimazole will be updated to reflect these safety issues.

An updated version of the Uniform Recall Procedure for Therapeutic Goods (URPTG) has been introduced by the Therapeutic Goods Administration (TGA). Major changes to the URPTG that are now in effect include:
- The introduction of two new recall actions; and
- Product defect correction. This term replaces ‘recall for product correction’ and is undertaken to correct a specific or potential deficiency. In some cases, the therapeutic good can continue to be used until a permanent correction is implemented. For example, pharmacies may be asked to replace plain bottle closures with child-resistant caps while awaiting the supply of new stock with the appropriate closure.
- Product defect alert. This is used to raise awareness of concerns regarding safety, quality, or performance while describing actions that may be taken to mitigate risks. This type of alert may be issued when discontinuation of treatment is considered a greater risk than continued use of the affected product. It is generally reserved for critical therapeutic goods that do not have an alternative and for which a recall action would result in a medicine shortage or interruption of patient treatment. A product defect alert may be followed by a recall once unaffected or alternative products become available.
- Introduction of one new type of non-recall action.
- A quarantine may be initiated when a defect is identified in released goods that has the potential to result in safety, efficacy, or performance issues. This suspends further supply and distribution of the goods while investigations are conducted, the results of which determine any further action that will be taken.
Further information on the other categories of recall and non-recall actions can be found at the TGA. HPS Pharmacies will continue to provide timely advice on drug recalls, safety, and supply issues via DrugAlert communications.